


1Department of Dermatology and Cutaneous Biology, Sidney Kimmel Medical College, and Jefferson Institute of Molecular Medicine, 2Genetics, Genomics and Cancer Biology PhD Program, Thomas Jefferson University, Philadelphia, PA, USA, and 3Department of Medical Genetics, School of Medicine, Tehran University of Medical Sciences, Tehran, Iran
The heritable forms of keratinization disorders, including various forms of ichthyosis and keratodermas, comprise a phenotypically heterogeneous group of diseases which can be divided into syndromic and non-syndromic forms. In the non-syndromic forms, the clinical manifestations are limited to the cutaneous structures while the syndromic ones are associated with a spectrum of extracutaneous manifestations. The inheritance in different families can be autosomal dominant, autosomal recessive or either X-linked dominant or recessive. Currently at least 67 distinct genes have been associated with different forms of ichthyosis. These genes can be grouped on the basis of their physiological involvement, including genes encoding structural components of epidermis, those involved in epidermal lipid metabolism, or those critical for cell-cell adhesion, and keratinocyte differentiation. This overview highlights some of the recent progress made in understanding the molecular genetics of keratinization disorders, and presents selected, recently characterized cases as representative of different forms of heritable ichthyosis.
Key words: autosomal recessive congenital ichthyosis; ichthyosis; non-alcoholic fatty liver disease; non-syndromic ichthyosis; syndromic ichthyosis.
Accepted Feb 12, 2020; Epub ahead of print Mar 9, 2020
Acta Derm Venereol 2020; 100: adv00095.
Corr: Jouni Uitto, MD, PhD, Department of Dermatology and Cutaneous Biology, Sidney Kimmel Medical College at Thomas Jefferson University, 233 S. 10th Street, Suite 450 BLSB, Philadelphia, PA 19107, USA. E-mail: Jouni.Uitto@jefferson.edu
Patients with ichthyosis manifest with dry and scaly skin, with considerable phenotypic variability. The heritable forms of ichthyosis are associated with mutations in over 60 different genes which encode proteins critical for normal physiological function of the skin. This overview highlights some of the new findings in the genetics of heritable forms of ichthyosis and emphasizes the connection of skin findings to extracutaneous manifestations, in some forms of the syndromic ichthyosis. The presentation also emphasizes the importance of determining the specific mutations in the underlying genes, which allows subclassification of the patients into distinct categories, with the capability to prognosticate the severity and the overall outcome of the disease in general terms. The knowledge of mutations in specific genes is also required for application of allele-specific therapies being developed for this group of disorders currently without specific treatments.
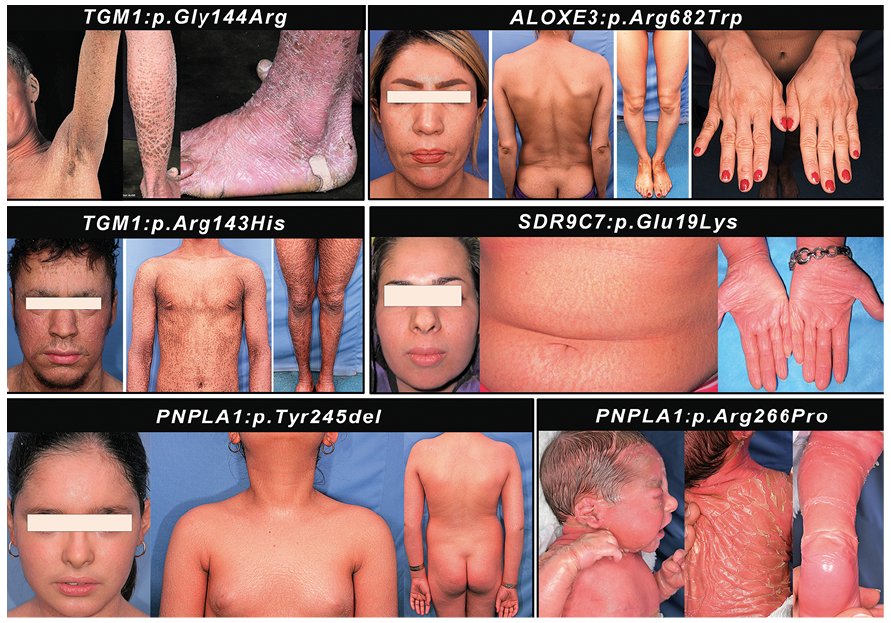
The heritable forms of ichthyosis, also known as generalized Mendelian disorders of cornification (MeDOC), comprise a heterogeneous group of diseases caused by mutations in a number of genes that are critical for development and maintenance of physiologic barrier at the outer layer of epidermis (1, 2). Clinically, these disorders manifest with scaling and hyperkeratosis of varying degrees (Fig. 1). The pathologic findings in the patients are either limited to the cutaneous structures or are associated with extracutaneous manifestations. Thus, ichthyosis can be divided into two broad categories, the non-syndromic and the syndromic forms. These disorders are present usually at birth or are diagnosed shortly thereafter, but the progression of the disease and the eventual outcome of severity can be highly variable. In the most severe forms, such as the Harlequin ichthyosis (HI), the affected children often die during the early neonatal period while at the other end of the spectrum of severity, such as ichthyosis vulgaris (IV), the manifestations can be relatively mild, the onset of manifestations may occur later in life and the spectrum may represent a continuum with physiologically present dry skin. The longevity of the IV patients is rarely affected by the disease. Also, in some cases the scaling and hyperkeratosis present at birth can be self-healing within a few months’ timeframe, as in so-called self-improving collodion ichthyosis (1–6).
Fig. 1. Phenotypic variability in patients with autosomal recessive congenital ichthyosis associated with defects in different genes. Note the spectrum of severity in association of specific mutations indicated.
Various forms of ichthyosis were initially classified on the basis of predominant clinical manifestations, and early on, different subtypes often carried eponyms of the authors of the original descriptions. More recently, it has been recognized that clinical heterogeneity, coupled with the variable mode of inheritance, i.e., autosomal dominant, autosomal recessive, or X-linked dominant or recessive, largely reflects the genetic heterogeneity, and currently as many as 67 distinct genes have been shown to be associated with different forms of ichthyosis. In addition, there are as many as 28 mutant genes associated with palmoplantar keratodermas. Grouping of the genes based on their involvement in distinct biological pathways required for physiological differentiation of keratinocytes and maintenance of the epidermal barrier has resulted in a more granular classification which allows refined diagnostics and nuanced prognostication in general terms (2). In such classification, specific subgroups of syndromic and non-syndromic forms of ichthyosis can be recognized, based on identification of mutations in genes encoding structural components of epidermis, involved in epidermal lipid metabolism or critical for cell-cell adhesion and keratinocyte differentiation as well as homeostasis, essential for formation of functional stratum corneum with uncompromised barrier function.
As in case of most heritable disorders with extensive genetic heterogeneity the identification of mutations in different forms of ichthyosis was initially based on PCR amplification of exons and flanking intronic sequences in candidate genes identified by clinical observations or by immunofluorescent and ultrastructural examination of epidermis. However, with expanding number of candidate genes associated with keratinization disorders, this approach has proven time-consuming and expensive. The PCR-based approaches are rapidly being replaced by next-generation sequencing (NGS) techniques, including sequencing arrays simultaneously targeting multiple disease-associated genes or the use of whole exome sequencing (WES) and whole genome sequencing (WGS) (7–9). These approaches are assisted by genome-wide tools, including homozygosity mapping (HM) and transcriptome profiling by RNA-seq, which facilitate identification and verification of pathogenic mutations in affected families (10–13).
A subgroup of non-syndromic forms of ichthyosis, autosomal recessive congenital ichthyosis (ARCI), is clinically divided into different subcategories: (a) Harlequin ichthyosis (HI), (b) lamellar ichthyosis (LI), and (c) congenital ichthyosiform erythroderma (CIE). HI is a rare and often severe form of ARCI caused by mutations in the ABCA12, while LI and CIE, with partially overlapping phenotypes, have been associated with mutations in a total of 14 distinct genes, many of them involved in lipid metabolism and essential for formation of functional stratum corneum. The different genetic subtypes were initially defined by the genomic locations of the corresponding genetic loci (ARCI1-17). The 4th subgroup of ARCI comprises phenotypically variable forms of ichthyosis which can manifest with marked hyperkeratosis at birth, but significant spontaneous improvement during infancy can result in a relatively mild disease in the adulthood (1–5, 14). This group, known as pleomorphic ichthyosis (14), consists of clinically distinct conditions, such as self-improving collodion ichthyosis, ichthyosis prematurity syndrome and congenital ichthyosis with fine/mild scaling (5, 7, 8). Bathing-suit ichthyosis, also a pleomorphic ichthyosis, is a condition in which arms and legs are not affected by abnormal keratinization (15). In addition, clinically defined forms of ichthyosis include peeling skin syndrome, erythrokeratoderma variabilis (EKV), loricrin keratoderma and congenital reticular ichthyosiform erythroderma. Ichthyosis is often associated with development of palmoplantar keratoderma, a heterogeneous group of disorders which can also present as distinct clinical entities without ichthyosis (see Thomas & O’Toole, 16).
In a recent study by us, the molecular basis of a total of 125 families with diagnostic features of ARCI was probed by a NGS array targeting 38 genes that were at the time known to be associated with different forms of ichthyosis (17). This approach, assisted by homozygosity mapping in this cohort which was characterized by high degree of consanguinity, followed by whole transcriptome analysis by RNA-Seq, identified definitive pathogenic/likely pathogenic mutations in approximately 85% of the families (for examples, see Fig. 2). Similar results have been reported in studies examining regional cohorts of families with ARCI, including patients in Czech Republic, England, Israel, Italy, Turkey and the Scandinavian countries (17–22). While some differences in the prevalence of mutations in different genes were observed in different cohorts, mutations in the TGM1 gene encoding transglutaminase 1, as well as in a number of genes involved in lipid metabolism (ABCA12, ABHD5, ALOX12B, ALOXE3, CERS3 and PNPLA1) were frequently encountered (23, 24). In some populations, PNPLA1 and CERS3 mutations are very rare while in others, particularly those with high degree of customary consanguinity, the proportion of these two genes was relatively high (25–29). Interestingly, while ABCA12 was initially associated with severe, often lethal HI, mutations in this gene can also be encountered in LI and CIE (30). It should be noted that many of the mutations found in different genes are private and population-specific, em-phasizing the importance of ethnic-based molecular diag-nostics when assessing its impact on the public health in different countries and geographic regions.
Fig. 2. Utility of RNA-based sequencing in identification and confirmation of a homozygous synonymous/splice-site mutation in TGM1 (Patient-1) and a non-canonical splicing variant in PNPLA1 in Patient-2. (a1) The proband, a female at neonate age (left panel) and at 3 years of age (right panel) showing extensive scaling and hyperkeratosis, harboring a mutation in TGM1. (a2) A 4-year-old female with generalized fine scaling and hyperlinearity of face harboring a mutation in PNPLA1. (b1) Screen shot of the genomic sequence visualized by the Integrative Genomics Viewer demonstrating a homozygous mutation of TGM1: c.1491G>A at the border of exon10/intron 10 (upper panel). Sashimi plot of RNA-Seq reveals that this homozygous synonymous mutation results in aberrant splicing and partial intron retention (lower panel, red), as compared to splicing in control RNA (blue). (b2) Screen shot of the genomic sequence visualized by the Integrative Genomics Viewer demonstrating a homozygous intronic mutation of PNPLA1: IVS1, c.205+5G>A (upper panel). Sashimi plot of RNA-Seq reveals that the homozygous non-canonical mutation in intron 1 results in the absence of PNPLA1 transcript (lower panel, red) as a result of possible loss of promoter and/or nonsense-mediated decay, as compared to an age- and sex- matched control (blue). (c1) Mean average-plot showing reduction of TGM1 gene expression (left panel) in Patient 1 (fold change: –1.4), and (c2) reduction of PNPLA1 gene expression (right panel) in Patient 2 (fold change: –4.5) in comparison to control. (Modified from ref. 17, with permission).
The most recent discoveries of ARCI-associated genes include SDR9C7 and SULT2B1. Initially described in 2016 in affected members of 3 consanguineous Lebanese families with congenital ichthyosis, SDR9C7 was mapped to chromosome 12q13-q14 (ARCI13) (29). This gene encodes short-chain dehydrogenase/reductase family 9C member 7 (SDR9C7) and is highly expressed in the granular and cornified layers of the epidermis. Subsequently, a 1-bp duplication co-segregated in a Turkish family was discovered, and as many as 14 mutations have now been reported in populations in Austria, Denmark, France, Germany, Iran, Japan, Sweden, Turkey and the United Kingdom (17, 31–34). Of note, presence of persistent fungal infection was frequently observed in these patients, suggesting that the functional SDR9C7 may physiologically provide protection against such infections.
Mutations in another gene, SULT2B1, were initially described in 2017 both in homozygous and compound heterozygous state, mapping to 19q13.3 (ARCI14) in 6 patients from 3 unrelated families (35). Subsequently, two missense mutations in two unrelated families were reported by us (Fig. 3) (17). The SULT2B1 gene encodes a sulfotransferase family cytosolic 2B member 1, expressed in the stratum granulosum-stratum corneum junction in the epidermis.
Fig. 3. Utility of homozygosity mapping in gene discovery in a consanguineous family with a SULT2B1 mutations. (a) The proband, a 32-year-old woman, demonstrated extensive scaling present since birth and hirsutism due to mutations in SULT2B1. (b) Pedigree of the three affected individuals with first cousin parents. Histopathology of the proband’s skin shows characteristic features of LI. (c) Autosome wide homozygosity mapping identified a region of homozygosity present in all 3 affected individuals (IV-1, 3, 4), but not in their parents (III-1, 2), on chromosome 19. This region harbored the locus for the SULT2B1 gene, and Sanger sequencing identified a homozygous mutation c.232G>A, p.Glu78Lys, as shown in (b). (Modified from ref. 17, with permission).
While the consequences of the mutations in non-syndromic forms of ichthyosis are limited to the skin, a number of cases with cutaneous keratinization disorder in association with extracutaneous manifestations have been reported (1, 2, 36). In such conditions, the pathway disrupted by the mutations in specific genes have consequences not only in the homeostasis of epidermis but also in a number of other tissues. As examples of such conditions serve two syndromes, neonatal ichthyosis associated with sclerosing cholangitis (NISCH) and Chanarin-Dorfman syndrome (CDS), in which in addition to skin, liver can be affected (37, 38). The NISCH syndrome is initially diagnosed with relatively mild ichthyosis at birth, and histology reveals thickening of stratum corneum at the outer layer of the epidermis (Fig. 4). Extracutaneous manifestations include hypotrichosis, scarring alopecia, hypodontia and enamel hypoplasia, but a critical element of this syndrome is the involvement of liver with sclerosing cholangitis diagnosed later in life on the basis of hepatomegaly and elevated serum levels of liver enzymes. Mutations in the CLDN1, which encodes the tight junction protein claudin-1, has been reported in a limited number of patients with NISCH syndrome (Fig. 4). Thus, congenital presentation of ichthyotic skin lesions together with mutations in CLDN1 as the molecular confirmation of NISCH syndrome can predict the development of sclerosing cholangitis and liver abnormalities.
Fig. 4. Clinical features and mutation detection in a consanguineous family with NISCH syndrome with claudin-1 deficiency. (a) The patients with fine scaly ichthyosis and alopecia, oligodontia and enamel hypoplasia. (b) Family pedigree of the affected individuals with consanguineous parents. (c) Gene-targeted next-generation sequencing identified an ultra-rare homozygous p.Tyr159Ter mutation in the patients, verified by Sanger sequencing, the parents being heterozygous carriers (red arrows). (d) Positions of the novel p.Tyr159Ter mutation (red) and those previously reported in the CLDN1 gene (black). NISCH, Neonatal ichthyosis associated with sclerosing cholangitis. (Modified from ref. 37, with permission).
In addition to NISCH syndrome, other forms of ichthyosis are associated with liver involvement. An example of such conditions is Chanarin-Dorfman syndrome (CDS) characterized by hepatomegaly and hepatic steatosis, in association with ichthyosis which is readily recognizable during early years of life due to mutations in ABHD5 (Fig. 5). Full-blown CDS with skin and liver findings is inherited in an autosomal recessive fashion due to loss-of-function mutations in ABHD5 (39, 40). The liver involvement often progresses to hepatic steatosis, liver fibrosis and cirrhosis, and may necessitate liver transplant.
Fig. 5. Pedigree structures and clinical findings in NAFLD families with ABHD5 mutations. (a) Family 1 is of nonconsanguineous Italian ancestry with a monoallelic mutation in ABHD5. Families 2–7 of Iranian ancestry with Chanarin-Dorfman syndrome (CDS) show extensive consanguinity. Heterozygous carriers (+/−) show evidence of NAFLD and/or dyslipidemia and type 2 diabetes mellitus, and patients with biallelic mutations (+/+) manifest with CDS with neonatal ichthyosis and NAFLD. Individuals labeled with § are considered obligatory carriers of the mutation. For the presence of clinical manifestations in individuals tested, see the color code. (b) Sanger sequencing of mutations in Families 1–6. The mutation of Family 7, ABHD5:c.560_578 del19 was published previously (61). (c) Positions of the distinct mutations along the ABHD5 consisting of 7 exons drawn to scale; the introns are not in scale. (d) Presence of lipid droplets (red) in leukocytes from control (upper panels), a heterozygous carrier (middle panel), and a homozygous individual (lower panel) after incubation without (-OA, left) or with 200 µM oleic acid (+OA, right). The lipid content was quantitated by assay of the pixel density of Oil red O and DAPI stained cells (bar graph). The values represent the mean ± SD of 105–125 cells for each sample. CDS, Chanarin-Dorfman syndrome; NAFLD, non-alcoholic fatty liver disease; OA, oleic acid; UTR, untranslated region. (Adapted from ref. 40, with permission).
An intriguing genetic constellation was recently recognized in heterozygous carriers of ABHD5 mutations in CDS families (40). Specifically these individuals had diagnostic features of dyslipidemia and non-alcoholic fatty liver disease (NAFLD), a multifactorial condition and the most common liver disease worldwide, affecting up to one-third of the Western populations (41, 42). The monoallelic loss-of-function mutations, identified in consanguineous families with CDS, resulted in NAFLD in an autosomal dominant inheritance pattern, which was confirmed in a large multi-generation family without consanguinity and with no individuals with biallelic mutations (40). These patients with the heritable form of NAFLD and/or dyslipidemia demonstrated complete clinical expression after the 4th decade of life, and the prevalence of ABHD5-associated NAFLD was estimated to be 1:1,137 individuals in general populations (40). Thus, mutations in ABHD5, which is involved in neutral lipid metabolism, emphasize the pathogenic role of lipid disorders both in NAFLD and in some forms of ichthyosis.
Recent independent studies corroborated the role of CGI-58 (encoded by ABHD5) and its partners, such as adiponutrin (encoded by PNPLA3) and ATGL (encoded by PNPLA2), in the pathogenesis of NAFLD. First, Romeo et al. (43) carried out a genome-wide association study (GWAS) of 9,229 individuals with NAFLD, and they found that a common SNP (rs738409[G]), encoding p.I148M in PNPLA3, was strongly associated with increased hepatic fat content (43). These observations have been supported by other studies. For example, Wang et al. showed a direct protein-protein interaction of CGI-58 and adiponutrin (44). In addition, they showed that normal PNPLA3 overexpression does not enhance lipid accumulation in primary hepatocytes derived from liver-specific Abhd5-knockout mice, thus again suggesting that PNPLA3 mediates ABHD5-dependent liver steatosis. Finally, Yang et al. showed that PNPLA3-I148M allele product in carriers attaches to and sequesters CGI-58 preventing its association with ATGL (Adipose Triglyceride Lipase) in a competitive inhibition fashion (45, 46). ATGL, when associated and activated by CGI-58, is required for the breakdown of triglycerides in the liver and adipose tissue. Thus, in the absence of ATGL available for binding to CGI-58, due to sequestration by PNPLA3-I148M in carriers, triglycerides accumulate in the liver providing a pathomechanistic explanation for hepatic steatosis in NAFLD (47).
In addition to claudin-1, a transmembrane protein in the tight junction complexes which regulates para-cellular permeability in the epidermis (see above), mutations in other genes involved in cell-cell communication have also been associated with aberrant keratinization phenotypes. One of such is GJB2 encoding connexin 26, previously shown to be mutated in KID syndrome and some forms of palmoplantar keratoderma (see Thomas & O’Toole, 16). Ichthyosis follicularis is a distinct keratinization disorder which has been reported in association with atrichia and photophobia resulting from mutations in the MBTPS2 gene (48). Recently, however, compound heterozygous mutations in the GJB2 gene were reported in a novel syndrome of ichthyosis follicularis, bilateral sensorineural hearing loss and punctate palmoplantar keratoderma (10) (Fig. 6). One of the mutations (p.Asn176Asp) was demonstrated to significantly reduce the cell-cell gap junction channel activity and to increase the non-junctional hemichannel activity of connexin 26 when tested in Xenopus oocyte expression system (10). This mutation, when associated with a common frameshift mutation in GJB2 (c.35delG; p.Gly12Valfs*2), frequently documented as a cause of sensorineural hearing loss, resulted in manifestations of this new syndrome, including ichthyosis follicularis phenotype. Collectively, these findings, coupled with previous reports on GJB2 associations with skin findings, attest to the complexity of clinical consequences of different mutations in GJB2.
Fig. 6. Example of a novel GJB2-associated syndromic form of ichthyosis. Pedigree structure, cutaneous features, histopathology, alteration of the magnetic field and mutations in GJB2 in families with autosomal recessive follicular hyperkeratosis, PPK, and bilateral sensorineural deafness. (a1, a2) Family pedigrees with autosomal recessive inheritance. (b1, b2) Histopathology of a skin lesion delineated in (b1) revealed a parakeratotic column of hyperkeratotic skin invaginating into epidermis. (c1 and c2). Palm of the proband (IV-1) with hyperkeratosis and accentuated creases which contain punctate pits. (d) Multiple discrete hyperkeratotic projections are centering on hair follicle in widespread distribution, including legs. (e and g). Filtering steps of whole exome sequencing data resulted in identification of compound heterozygous p.Asn176Asp and c.35delG mutations in GJB2, followed by Sanger sequencing confirmation. (f) Modeled structure of Cx26 gap junction channels in wild-type (left) and mutant p.Asn176Asp (right). Note the alteration in positive (blue) and negative (red) electrostatic potentials. (Modified from ref. 10, with permission).
A number of other genes contributing to cell-cell adhesion and communication have been associated with syndromic forms of keratinization disorders. For example, the desmosomal proteins JUP and DSP have both been associated with cardiomyopathy, but cutaneous manifestations, such as palmoplantar keratoderma or hair abnormalities, are highly variable depending on the mutation involved (49, 50). More recently, it was demonstrated that alterations in PERP, another component of desmosomes can cause an autosomal dominant Olmsted syndrome or autosomal recessive erythrokeratoderma (51). This observation underscores the genotypic and phenotypic heterogeneity of keratinization disorders: PERP mutations confer a spectrum of phenotypes ranging from severe periorificial plaques and palmoplantar keratoderma, as seen in patients with TRPV3 mutations (52), to varying degrees of erythrokeratoderma observed in patients with mutant GJB3, GJB4, and LOR genes (53–55).
The PERP gene consists of 3 exons encoding the p53/p63 tetraspan membrane protein that is expressed primarily in stratified epithelia (56, 57). Although the exact interacting partners of PERP are still unknown, its importance in cell-cell adhesion and epithelial integrity was implied by the observation that the majority (95%) of Perp knockout (–/–) mice died within 10 days of life due to blistering in the oral mucosa and skin, especially in areas of mechanical trauma, but also showed abnormal thickening of the epidermis (57). The 5% of Perp–/– mice that survived to adulthood had a significantly shorter lifespan compared to Perp+/– and wild-type mice and did not show a predisposition to spontaneous tumorigenesis despite evidence linking p53/p63 to Perp expression (56, 58). Nevertheless, with documentation of these cases with PERP mutations in humans, clinicians should be aware of this connection and monitor patients accordingly for potential evidence of carcinogenesis.
Erythrokeratoderma manifests with hyperkeratotic, often transient and migratory erythematous and figurate plaques with sharply demarcated borders typically developing in early childhood (Fig. 7). It has been historically divided into two main categories: (a) erythrokeratodermia variabilis et progressiva; and (b) progressive symmetric erythrokeratoderma. However, these two presentations are currently listed on the OMIM catalogue under a single disease entry (OMIM #133200). There are a number of other presentations with erythrokeratoderma (59). Ery-throkeratoderma can be inherited either in an autosomal dominant or an autosomal recessive pattern. The autosomal dominant forms have been associated with mutations in the gap junction-related genes (GJB2, GJB3, GJB4, and GJA1) as well as in LOR, encoding loricrin, a cornified envelope protein (53–55, 59). Autosomal recessive erythrokeratoderma has been associated with mutations in ABHD5, ELOVL4 and KDSR. More recently, the genotypic spectrum of erythrokeratoderma has been extended by application of NGS using ichthyosis-associated gene sequencing panels which identified mutations in addition to those previously identified genes, also in PNPLA1 in families with the autosomal recessive form of erythrokeratoderma (Fig. 7) (10). These studies provide evidence in support of the notion that erythrokeratoderma can be a manifestation associated with multiple types of ichthyosis with different gene defects (60). Consequently, erythrokeratoderma may not be a distinct genetic entity but rather a manifestation of multiple ichthyosis-related genetic diseases that can occur with or without a more typical ichthyosis presentation.
Fig. 7. Patients with ichthyosis and different genetic mutations presenting with erythrokeratoderma. (a) Patient 1, a 2-year-old female patient with erythrokeratoderma and a homozygous missense mutation in the ABHD5 gene consistent with Chanarin-Dorfman syndrome. (b) Patient 2, a 7-year-old male with erythrokeratoderma and generalized ichthyosis due to a missense mutation in PNPLA1. (c) Patient 3, a 12-year-old female with extensive erythrokeratoderma and large brown ichthyotic plaques on the face, consistent with autosomal recessive ichthyosis associated with mutations in PNPLA1. Sanger sequencing confirmed the out-of-frame deletion of 13 bp, shown in yellow (lower right panel) (Modified from ref. 57, with permission).
This update on recent advances in our understanding the molecular basis of heritable keratinization disorders highlights the tremendous variability, both phenotypic and genotypic, in this group of disorders. The knowledge of the mutant genes and of specific mutations can be used to confirm the diagnosis with subclassification, allows determination of the mode of inheritance, and provides information for prognostication, in general terms, of the severity and overall outcome of the disease. The mutation detection in large consanguineous families also allows identification of heterozygous carriers which can be coupled with genetic counseling for the risk of recurrence in the extended family. The mutations form the basis for prenatal testing and preimplantation genetic diagnosis. Finally, the knowledge of the specific mutations is a prerequisite for allele-specific treatments currently being developed for this group of complex disorders without specific treatment modalities.
Jason Park and Madeleine Kilimnik, medical students in the laboratory of Dr. Uitto, contributed to the review of the literature. The authors thank Carol Kelly for manuscript preparation.
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize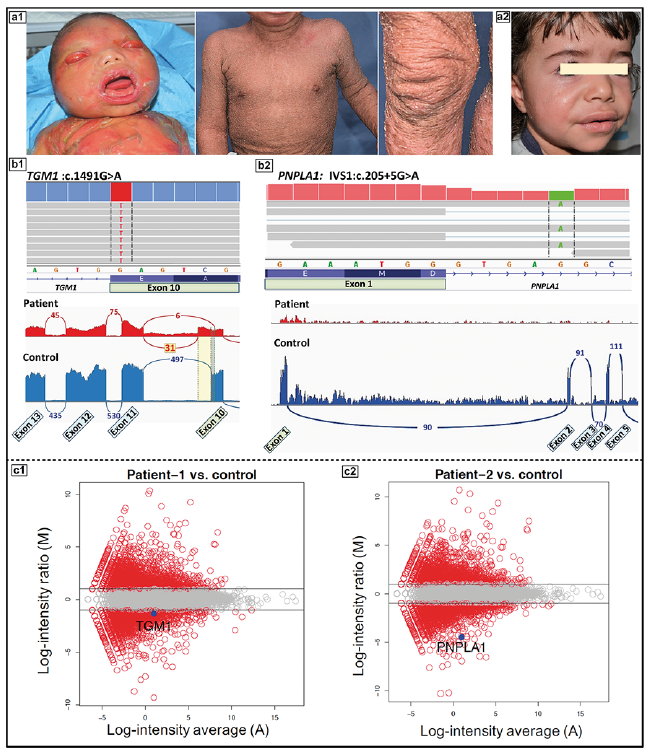 Click to show fullsize
Click to show fullsize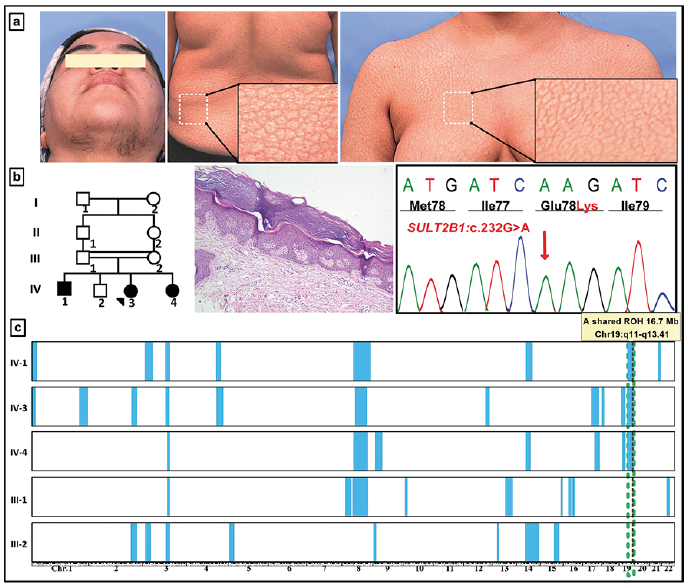 Click to show fullsize
Click to show fullsize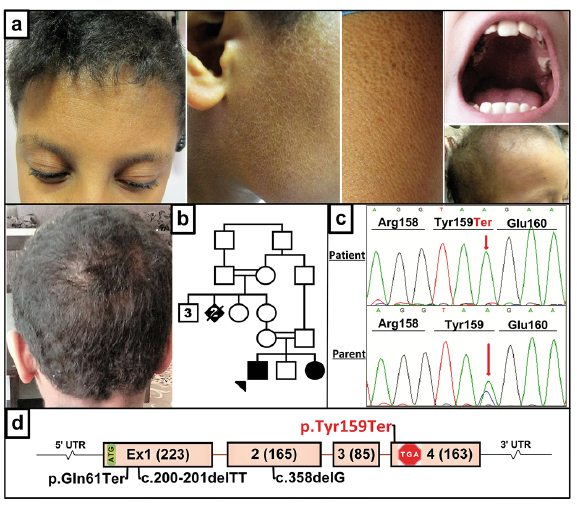 Click to show fullsize
Click to show fullsize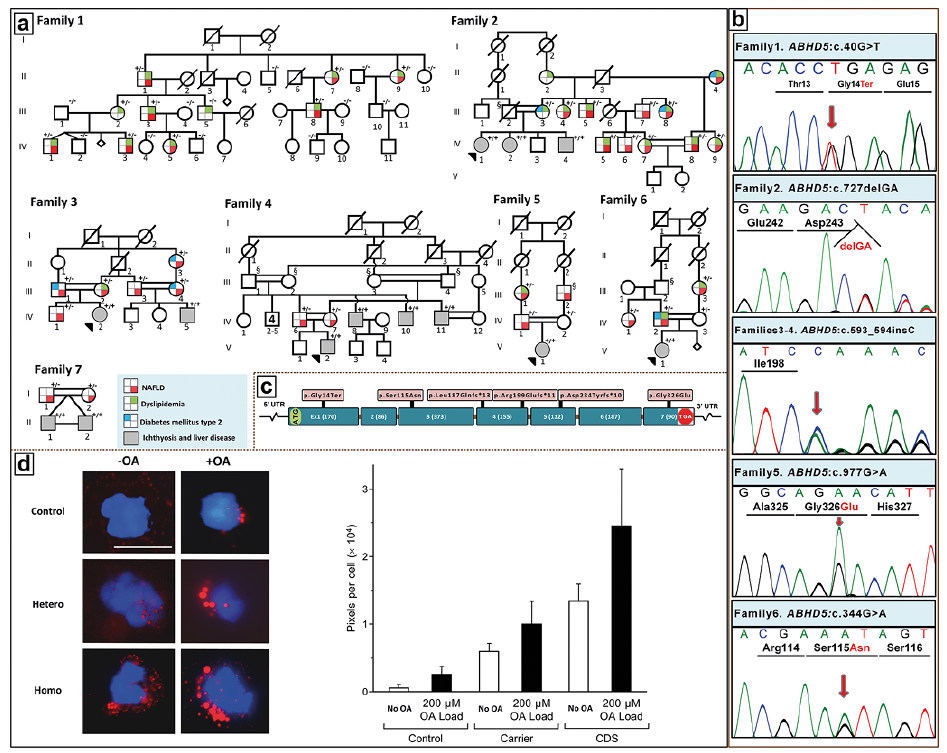 Click to show fullsize
Click to show fullsize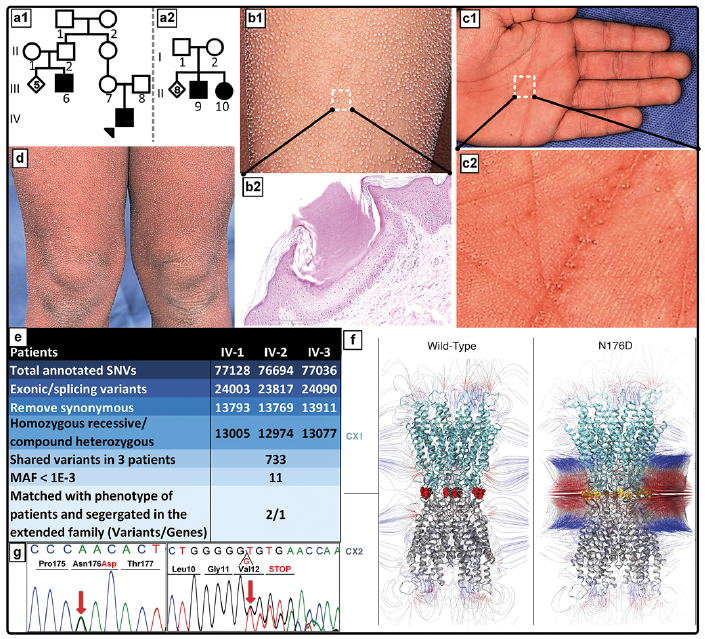 Click to show fullsize
Click to show fullsize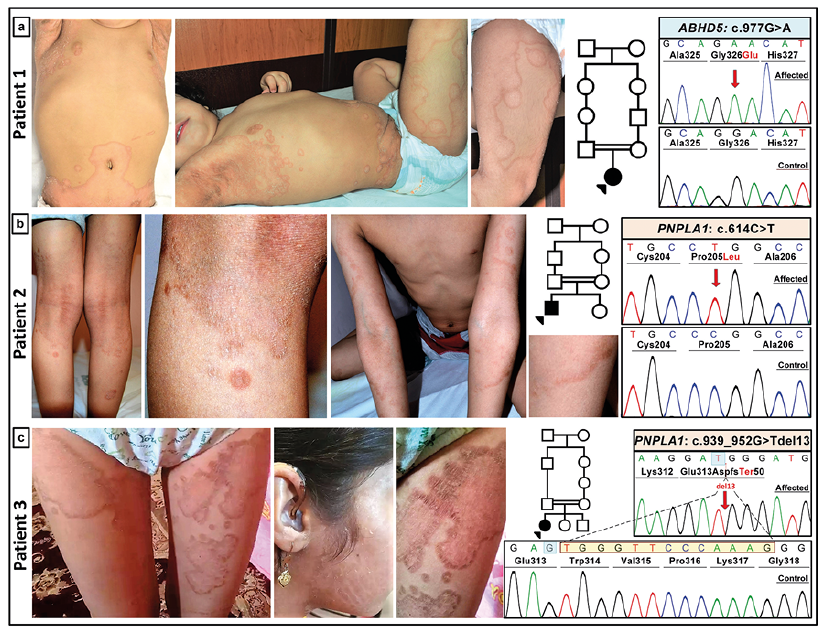 Click to show fullsize
Click to show fullsize